- 481
- 产品价格:面议
- 发货地址:湖北武汉江汉区 包装说明:不限
- 产品数量:9999.00 个产品规格:不限
- 信息编号:242363962公司编号:14192389
- 蒋小姐 商务代表 微信 180071419..
- 进入店铺 在线咨询 QQ咨询 在线询价
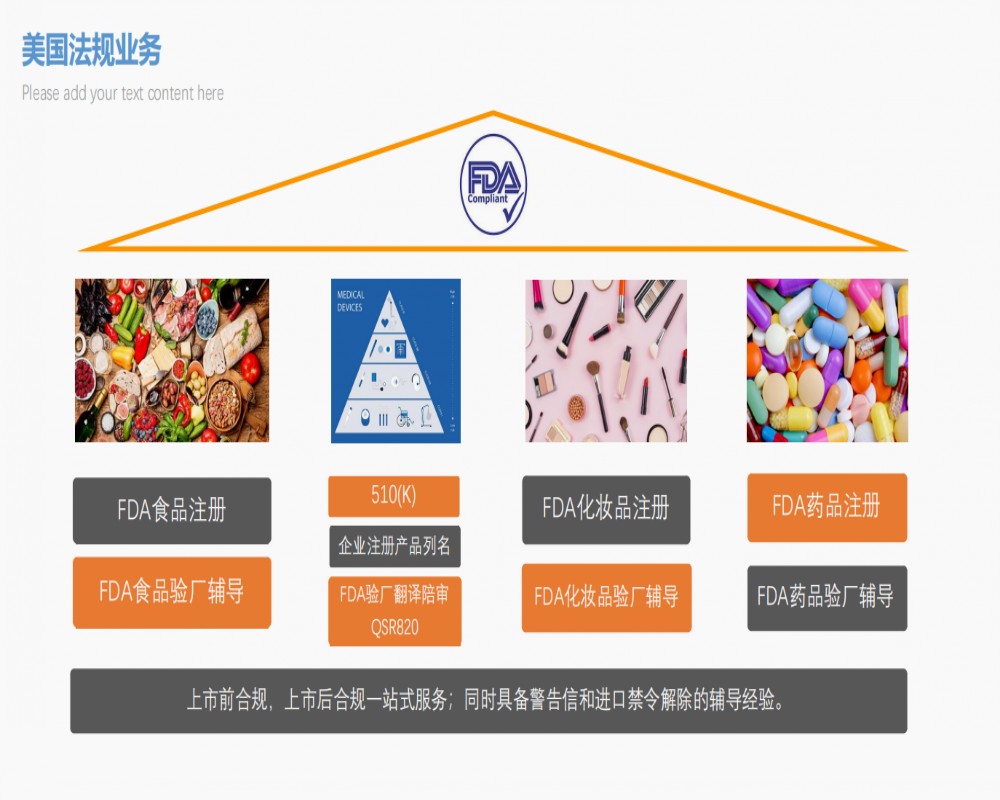
马鞍山医疗器械MDR CE认证周期 需要什么材料
- 相关产品:
ce认证周期
周期:4周左右,具体周期根据企业产品调整服务:一对一专业服务其他认证服务:IVDR CE/FDA 510K/FDA 验厂/UKCA/CFS/产品检测等认证内容:MDR CE服务服务范围:全国
MDR认证要则:欧洲医疗器械协调小组(MDCG)更新了一份问答文件,内容涉及与即将生效的医疗器械条例MDR和体外诊断医疗器械条例IVDR相关的公告机构和联合评估相关的合规性要求。MDR的主要变化:
1.扩大申请范围;
2.提出的新概念和设备定义;
3.优化医疗器械的分类;
4.提高设备的一般*性和性能要求;
5.加强对技术文件的要求;
6.设备上市后加强监督;
7.提高评估的相关要求;
8.建议建立和使用Eudamed数据库;
9.提出设备可追溯性(UDI);
10. NB的严格要求。
MDR法规下医疗器械CE认证;
1、产品性能检测
依据产品标准或者技术要求,对产品进行的全性能检测。
接故的板:材料,机械性能,表面质量,尺寸,静态四点弯曲,疲劳四点弯曲。
接骨螺钉:材料,机械性能,表面质量,尺寸,螺钉旋入旋出,扭转,自攻,静动态四点弯曲。
2、选样说明
一般选择具代表性,较差情况型号进行检测。如果一个型号无法覆盖,需测试2个或者多个型号。
3、测试机构
如果企业有设备有能力检测,可以企业自测。否则就找有资质的第三方测试。
MDR认证关键时间点:主要有三个时间点需注意,且不要混淆.
1)欧盟医疗器械新法规MDR (REGULATION EU 2017/745) 颁布时间:2017年5月。
法规规定:新的法规将替代原有的医疗器械指令 (MDD 93/42/EEC) 和有源植入性医疗器械指令(AIMDD 90/385/EEC)
2)MDR法规强制实施时间:2020年5月26号
法规规定:从2020年5月26号开始公告机构不能按照MDD颁发CE证书,目前I*及以上风险等级产品认证机构已不再受理MDD指令的认证申请。
3)MDD失效时间:2024年5月27号
2024年5月27号起,企业持有的MDD指令的CE证书全部失效。
MDR法规下所谓的CE证书并不是,因为不是法规要求;
MDR下合规的是:
1)技术文件是否满足DRM的要求;
2)欧代是否按照MDR的要求进行了器械注册。
MDR法规对于普通一类没有提出认证要求;
MDR法规下,普通一类也不需要公告机构评审。
我们公司本着“客户至上,诚信至上”的原则,秉持“管理创造价值、服务提升优势、品质至上、服务至优"的发展理念,欢迎新老客户参观与咨询。
联系电话是17717017570, 主要经营上海沙格医疗科技有限公司武汉分公司(咨询电话:18007141921)是一家QSR820验厂,主营:IVDR CE认证、MDR CE认证、英国UKCA认证、FDA产品测试、ISO13485体系认证等服务。十多年里,SUNGO已为**30多家上市公司和**制造商,合计5000多家企业提供过相关服务。。
单位注册资金单位注册资金人民币 100 万元以下。
{kind=link}